Dr. Lazar | Prof. Dr. Seideman | Dr. Samaras
We are LSS International
Dr. Lazar | Prof. Dr. Seideman | Dr. Samaras
We are LSS International
Our Global Leadership Team
Neurology | Neuroimmunology | Endocrinology
The photographs on this website were, of course, edited by our media agency — they made us look much better, but they did not create us.

Dr. Marius Lazar
New York City and Athens EU
Founder of LCG & LSS
Neurology
Psychiatry
Neurodermatology

Prof. Dr. Carolina Diamandis
Athens Nicosia, EU
Medical Director of LCG
(Research on ex juvantibus therapies)
Military Physician (ret.)

Prof. Dr. David Seideman
Athens, Tel Aviv and Denver
Medical Director of LSS
Neurology
Neuroimmunology
Neuroendocrinology

Prof. Dr. Dr. Helena Samaras
Cyprus, Athens, EU, U.S.A.
Deputy Medical Director (LSS)
Neurology & Endocrinology
LSS & LCG International
Military Physician (ret.)

Dr. Alexandros Balaskas
Athens
Head of the team of assistant doctors team one (Neurology, Endocrinology)
Assistants
- Dr. Schmidt-Christidis
- Dr. Sofia Makri
- Dr. Georgios Papadopoulos
- Dr. Nicholas Antonakis

Dres. Iias & George Samaras
Cyprus (entire Island)
The Samaras brothers
Neurometabolic conditions
Metabolic diseases
Autoimmune diseases
Greek only

Dr. Olga Ivanova
Athens, Bucharest, Sibiu
Head of the team of assistant doctors team two (Neurology)
Assistants
- Dr. Olaf W. Sørensen
- Dr. Kostas Karopoulos
- Dr. Norbert Bogdanski
- Dr. Sven Olsen

Dr. Jonathan Feldman
German-East-Belgium
General Medicine
Orphan Diseases
Dermatology
German Speaker (Belgium)

Dr. Lucas Smith
New York City
Endocrinology
General Medicine

Dr. Merab Joseph
Brazil, Georgia, NYC
Senior staff member
Cardiology

Dr. Alexander Ivanov
Pretoria
Senior staff member
Endocrinologist
Mitochondriopathies
General Medicine

Dr. Ali Asgari
Far East
Senior Staff Member for pharmacology
Specialist for off-label and innovative drug use

Eric Stone. Ph.D.
New York City and Denver
Curator of LCG Research at Harvard Dataverse

Walter Seideman, MD
New York City and Cyprus
CEO of the LCG Group
Medical examinations for telemedicine patients
Possible thanks to our international partners.

Laboratory tests
All state of the art laboratory tests are available in more than 30 countries.

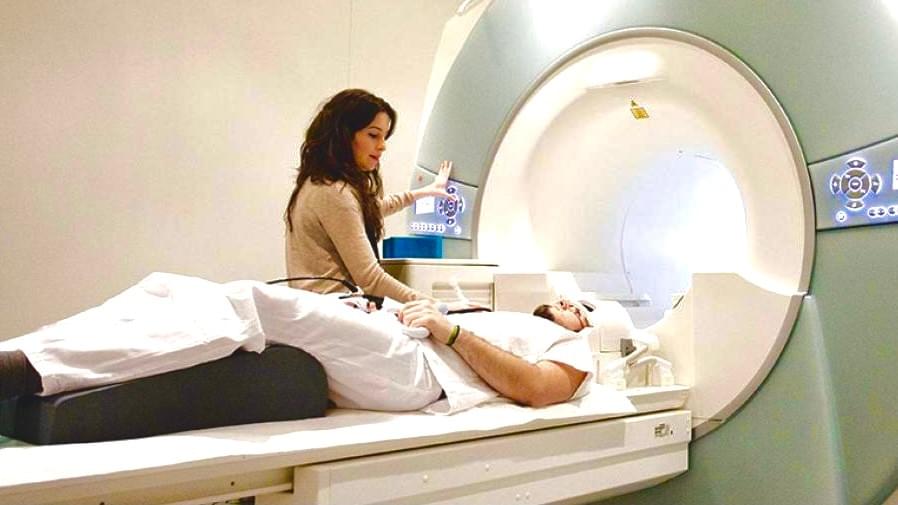
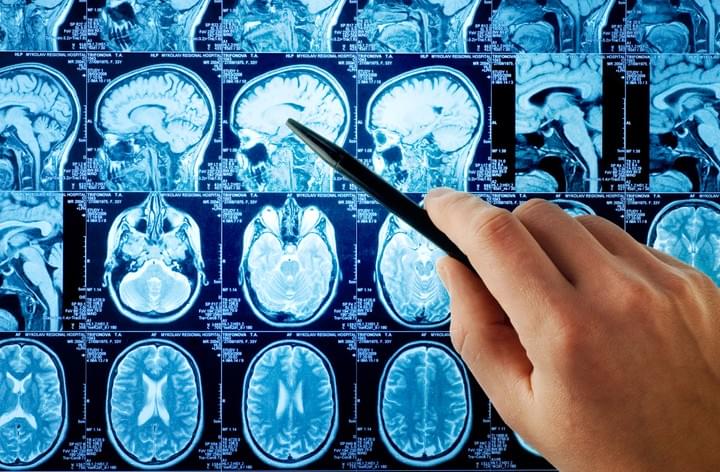
MRI, CT, TCS
Our partner practices and clinics perform examinations with MRI, CT and TCS (sonography of the brain). Scintigraphy is also still possible but less common than in the past. Due a three-step diagnostic procedure you will get an unusually high level of safety.

Neurological exams
All standard neurological tests are being provided by carefully selected partners in over 50 countries.
LCG Greece Research
We share our experiences as clinicians for clinicians with LCG Greece Research
Working with rare diseases means learning, an ongoing process.
We share clinical observations as quickly as possible, bypassing lengthy academic discussions.

We are proud EU-Europeans, dear friends of the United Kingdom, the Jewish nation, as well as blessed with close ties to the United States. In whatever we do, we try to represent the best the EU has to offer: compassion, innovation, and a little bit of a creative chaos.

OpenAIRE
We are a proud member of open AIRE (Open Access Infrastructure for Research in Europe) is a European initiative aimed at promoting open access to research outputs and facilitating the discovery and reuse of research information. It provides a platform for researchers, institutions, funders, and policy makers to discover and access the full text of research articles, datasets, and other research outputs, as well as information about projects, funding, and collaborations.
OpenAIRE offers a variety of services and tools for researchers, including a discovery portal for accessing open access research outputs, support for data management and sharing, and integration with research information systems. The initiative also provides guidance and support for researchers and institutions to comply with open access mandates and funding requirements.
OpenAIRE aims to make research outputs more discoverable and accessible, enabling researchers to build on the work of others and facilitate collaboration and innovation in research. It plays a key role in promoting open science and advancing the transition to a more open, collaborative, and transparent research ecosystem.
The Doctor will see you now.
Here you are taken seriously.

How Lazar | Seideman | Smith works
Clear rules.
Our top priority is to treat you as individually as your condition requires. Remote treatments (video telemedicine) in the strict sense are performed, but only in well-defined cases. Highly qualified colleagues in over 20 countries perform those physical examinations for us which are not possible via video conference and telemedicine.

Prudence, vigilance and diligence.
Remain humble to what you don’t know.
Since no one is omniscient, being well-networked makes a big difference. Dr. Lazar has always made it a point to network with as many colleagues as possible. After all, there's nothing worse than a doctor who thinks he knows everything. Therefore “better safe than sorry” is his credo.
Dr. Lazar puts great emphasis on continuously educating himself and sharing his knowledge. That's why he also contributed pro bono as an editor to DocCheck's medical encyclopedia, among other projects.
Diagnosing and treating rare diseases is more important than ever. Thousands of rare diseases exist, up to about seven thousand rare diseases have already been described scientifically, and more are being identified on an ongoing basis. Yet they remain mostly unknown in day-to-day medical practice. Medical and scientific knowledge about rare diseases is far from sufficient in the medical profession. The subject area of rare diseases has long been neglected by physicians, politicians and scientists, so that neither appropriate health policies nor scientific initiatives existed.
For most rare diseases there is no efficient therapy, nevertheless appropriate interventions can improve the quality of life and also increase life expectancy. Some impressive progress has been made in the treatment of certain diseases, which should motivate us to intensify research efforts.
Patients with rare diseases mostly face similar problems. Difficulties exist mainly in terms of diagnosis, availability of relevant information and referral to appropriate specialists. Equally important is access to qualified specialist facilities, general social and medical support, effective cooperation between hospitals and general practitioners, professional and social integration or maintaining independence.
Those affected often face psychological, social, economic or cultural problems. Many patients do not receive a diagnosis because the lack of sufficient scientific and medical knowledge means that a rare disease is often not recognized. It is precisely this group of patients that is exposed to the greatest pressure of suffering, as they are denied access to adequate support.

We are first and foremost human beings.
And care about our patients, more than usual.
Those with a rare disease who are patients of Lazar, Seideman & Smith see their attending specialist every 10 to 14 days. In addition, a team of assistant physicians with a variety of specialties is available 24 hours a day for our long-term patients. Those who receive their treatment under the Luzia Healthcare u.n.a. social program are not billed for treatment by our assistant doctors. Or, to put it briefly: we take good care of our patients.

Rare diseases are a challenge
What we are confronted with.
Orphan diseases, also known as rare diseases, are defined as conditions that affect fewer than 200,000 people in the United States. There are over 7,000 rare diseases affecting approximately 400 million people worldwide. Diagnosis and treatment of rare diseases face several challenges, including:
- Lack of Awareness: As the name suggests, these diseases are “rare” and as such, there is often limited awareness and understanding of these conditions among the general public and even healthcare professionals. This can lead to misdiagnosis and delayed treatment.
- Limited Research and Development: Due to the small patient population and limited financial incentives, there is often limited research and development into orphan diseases. This can lead to a lack of knowledge about the underlying mechanisms and treatments for these conditions.
- Lack of Diagnostic Tools: There is often a lack of diagnostic tools and tests for rare diseases, leading to difficulties in accurately diagnosing the condition.
- Difficulty in Finding Specialists: There may be only a few healthcare professionals who specialize in a particular rare disease, making it difficult for patients to access specialized care and treatment.
In conclusion, the low prevalence of orphan diseases makes it challenging to diagnose and treat them effectively. The limited research and development, lack of diagnostic tools, and difficulty in finding specialists all contribute to the difficulties faced in diagnosing and treating these conditions.
That's the reason why we do what we do.
EU-Prescriptions
In the EU, there is an initiative to facilitate the cross-border validity of medical prescriptions to support patient mobility within the EU. However, EU-prescriptions will remain on paper, not digital because the digital systems across different EU member states are currently quite varied and lack uniformity.
EU prescriptions, valid in all member statea, are expected to remain in paper form, rather than transitioning to digital, for the foreseeable future.
In practice, this means that paper prescriptions continue to play a crucial role, particularly for patients who receive treatment in one EU country but need to obtain medication in another EU country. The use of paper prescriptions in such cases allows pharmacists in the foreign country to more easily understand and dispense the medications as prescribed.
While the EU is working towards harmonizing digital health systems, including e-prescription systems, to improve compatibility and interoperability across different member states, paper prescriptions remain essential for cross-border healthcare within the EU until this harmonization is fully realized in the far future.

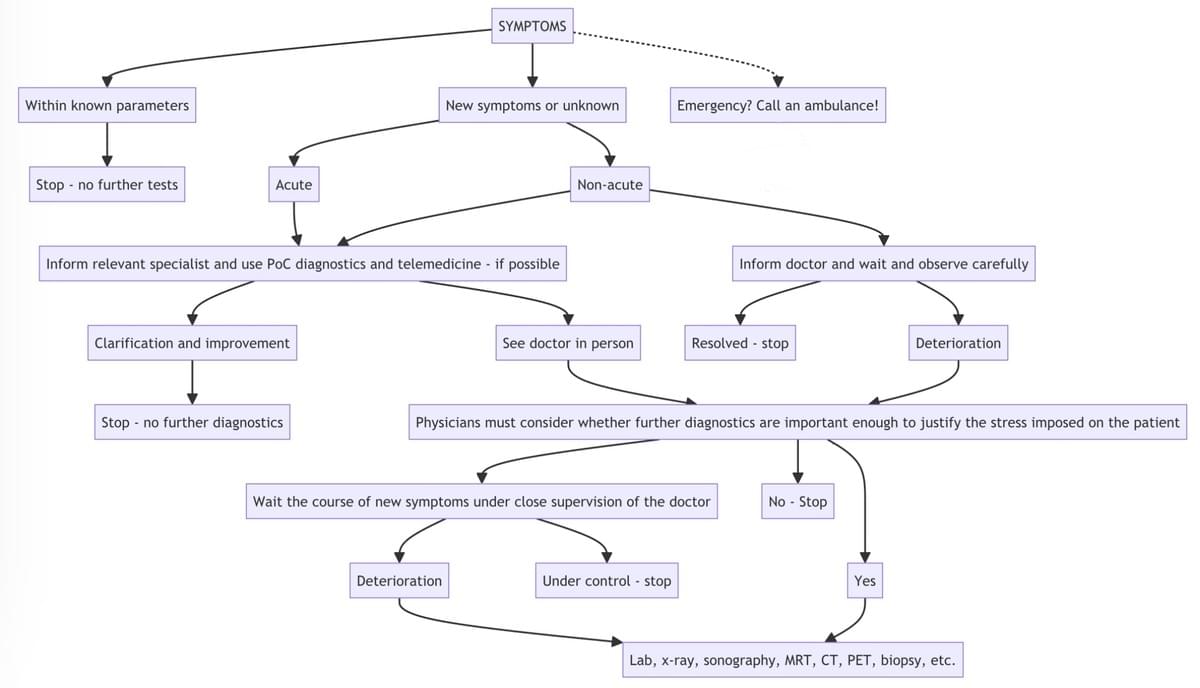
How to manage a rare disease
A course of action that we consider to be reasonable

Wir helfen auch auf Deutsch.
Und das hat einen guten Grund.
Als einzige hochspezialisierte Netzwerkpraxis für neurologieche, neuroendokrinologische und neurometabolische Erkrankungen, stellen wir konsiliarisch unser Wissen für Ärzte und Patienten zur Verfügung, und dies in der gesamten Europäischen Union. Herr Dr. Lazar und Prof. Dr. Seideman haben beide private enge Bindungen an Deutschland bzw. die deutsche Sprache, so dass das Deutsche sogar eine von mehreren Arbeitssprachen unseres Teams ist. Natürlich pflegen wir auch enge Kontakte zu ärztlichen Kollegen in Österreich, der Schweiz und sporadisch auch in Deutschland, um eine gute Versorgung dieser Patienten am Wohnort sicherstellen zu können. Wir nennen Ihnen gerne Fachärzte für Allgemeinmedizin, hausärztliche Internisten sowie Kollegen aus anderen Fachgebieten im deutschsprachigen Raum. Nehmen Sie einfach über das Formular oder telefonisch Kontakt mit uns auf.

Hausbesuche
Wir sind keine lokal fixe Praxis, sondern arbeiten in einem Netzwerk, das ganz die gesamte EU überspannt. Die leitenden Ärtze der LSS unterstützen lokale Kollegen mit ihrem Know-how. Hausbesuche werden in der Regel von erfahrenen hausärztlichen Kollegen in der EU durchgeführt.